Introduction
Cardiometabolic diseases, including atherosclerotic cardiovascular disease, stroke, type 2 diabetes, and non-alcoholic fatty liver disease, remain the leading causes of morbidity and mortality worldwide, imposing substantial clinical and economic burdens on aging populations. These conditions are tightly intertwined with aging biology, where chronic low-grade inflammation (“inflammaging”), endothelial dysfunction, a prothrombotic milieu, endoplasmic reticulum (ER) stress, and lipotoxicity act as convergent drivers of vascular and metabolic across the lifespan.
Despite major advances in pharmacotherapy, most notably statins, antihypertensives and antiplatelet agents, large cohorts continue to exhibit considerable residual cardiovascular risk, particularly those with metabolic syndrome, insulin resistance, and subclinical atherosclerosis. Even in individuals achieving guideline-directed targets for low-density lipoprotein cholesterol and blood pressure, persistent abnormalities in coagulation, microvascular function, and cellular stress pathways contribute to ongoing risk of major adverse cardiovascular events and progressive end-organ damage.
In this context, there is growing interest in evidence-informed nutraceuticals as adjunctive strategies aimed at complementary biological pathways not fully addressed by conventional therapies. Nattokinase, a food-derived fibrinolytic serine protease produced during fermentation of soybeans into natto, has been investigated for its capacity to enhance fibrin degradation, modulate homeostasis, and favorably influence vascular parameter. Tauroursodeoxycholic acid (TUDCA), a hydrophilic taurine-conjugated bile acid, has emerged from hepatology and neurology research as a cytoprotective molecule with applications in cholestatic liver disease, metabolic disorders, and neurodegenerative conditions through mitigation of ER stress, apoptopsis and inflammation.
Given the converging roles of thrombosis, endothelial injury, ER stress, and metabolic dysfunction in cardiometabolic disease and aging, a combined approach using nattokinase and TUDCA represents a biologically plausible strategy targeting both vascular and cellular stress axes. The objective of this review is to synthesize current mechanistic and clinical evidence for nattokinase and TUDCA individually, explore their theoretical synergy in cardiometabolic and aging-related pathways, and propose a research agenda to evaluate this combination for metabolic and vascular health in both clinical and preventive settings.
Nattokinase: Mechanism and Evidence
Nattokinase is a fibrinolytic serine protease produced by Bacillus subtilis var. natto during the fermentation of soybeans into natto, a traditional Japanese food. Commercial preparations are typically standardized in fibrinolytic units (FU), reflecting enzymatic activity rather than mass, with common supplement doses ranging from 2,000 to over 10,000 FU per day in clinical and observational studies. Human pharmacokinetic work suggests that nattokinase exhibits at least partial oral bioavailability, relative stability in the gastrointestinal tract, and a plasma half-life that appears longer than many other orally administered plant-based enzymes, enabling sustained fibrinolytic effects after single or repeated dosing [1,2].
Mechanistically, nattokinase exerts both direct and indirect fibrinolytic actions. In vitro and in vivo experiments demonstrate that the enzyme can directly degrade fibrin and fibrinogen, and also enhance fibrinolysis indirectly via activation of plasminogen and modulation of endogenous anticoagulant pathways. These activities translate into reductions in circulating fibrinogen and D‑dimer in some human studies, suggesting a net shift toward enhanced clot resolution without the same mechanism of action as conventional anticoagulants. Beyond fibrin degradation, nattokinase has been reported to exert antithrombotic and hemorheologic effects, including decreased whole‑blood viscosity and attenuation of platelet aggregation in small animal and human investigations, potentially improving microcirculatory flow and reducing thrombotic propensity [1].
Vascular and lipid parameters also appear to be modulated by nattokinase in longer‑term supplementation studies. An open‑label trial using high‑dose nattokinase (10,800 FU/day for 12 months) reported significant reductions in carotid intima‑media thickness and carotid plaque size, accompanied by improvements in lipid profile, with decreases in triglycerides and low‑density lipoprotein cholesterol and increases in high‑density lipoprotein cholesterol. These findings have been interpreted as consistent with anti‑atherosclerotic and plaque‑stabilizing effects, although the absence of large randomized, placebo‑controlled trials and the potential for selection and confounding biases warrant cautious interpretation [2].
Clinical data in blood pressure management further support a potential cardiometabolic role for nattokinase. Small randomized controlled trials in prehypertensive and stage 1 hypertensive individuals have shown modest but statistically significant reductions in systolic and diastolic blood pressure after several weeks to months of nattokinase supplementation compared with placebo, with some studies also noting concurrent improvements in biomarkers of coagulation and fibrinolysis. Observational and open‑label cohorts, often using higher doses and longer follow‑up, additionally describe regression of carotid atherosclerotic plaques and favourable shifts in lipid parameters, aligning with the mechanistic evidence on vascular and hemorheologic modulation [1,2].
From a safety perspective, nattokinase has generally been well tolerated in clinical studies, with few serious adverse events reported at commonly used doses. Nonetheless, the enzyme’s fibrinolytic and antithrombotic properties raise a theoretical and, in some case reports, observed risk of increased bleeding, particularly when used concomitantly with anticoagulant or antiplatelet medications, or in individuals with underlying coagulopathies. Consequently, cautious patient selection, avoidance in those with active bleeding or recent major surgery, and careful monitoring when combined with other agents affecting hemostasis are prudent until more robust safety data from large, well‑controlled trials become available [1].
TUDCA: Mechanism And Evidence
Tauroursodeoxycholic acid (TUDCA) is a hydrophilic taurine‑conjugated bile acid with greater aqueous solubility and a more favourable cytoprotective profile than many endogenous hydrophobic bile acids, which can be detergently cytotoxic at higher concentrations. Following oral administration, TUDCA is efficiently absorbed in the small intestine and enters the enterohepatic circulation, leading to preferential hepatic uptake and enrichment, while a fraction recirculates systemically to exert extrahepatic effects on metabolic tissues and the nervous system. This pharmacokinetic behaviour underpins its development as a hepatoprotective and neuroprotective agent and provides a mechanistic basis for its use in chronic cholestatic liver disease and emerging metabolic indications [3,4,5].
At the mechanistic level, TUDCA is widely characterized as a chemical chaperone that mitigates endoplasmic reticulum (ER) stress and modulates the unfolded protein response (UPR). In diverse cell types, TUDCA attenuates activation of canonical ER‑stress sensors, including PERK, IRE1, and ATF6, reduces downstream phosphorylation of eIF2α and expression of CHOP, and improves protein‑folding homeostasis, thereby preventing UPR‑driven apoptosis. These effects are complemented by mitochondrial protection: TUDCA preserves mitochondrial membrane potential, limits cytochrome c release, and shifts the balance of Bcl‑2 family proteins toward anti‑apoptotic signaling, with up‑regulation of BCL‑2 and down‑regulation of Bax, cleaved caspase‑3, and cleaved PARP reported in preclinical models [6,7,8].
Beyond its role in proteostasis, TUDCA exerts anti‑inflammatory and metabolic actions that are particularly relevant to obesity‑related and hepatic disease. In high‑fat diet models of non‑alcoholic fatty liver disease (NAFLD), oral TUDCA reduces hepatic steatosis, improves insulin resistance, and lowers fasting glucose and insulin, accompanied by down‑regulation of inflammatory cytokines and improvement in gut–liver axis parameters. Additional work in obese and type 2 diabetic mice shows that TUDCA normalizes glycemia, restores insulin sensitivity in liver and peripheral tissues, and may enhance β‑cell function via modulation of ER stress and bile‑acid–sensitive receptors such as TGR5. Collectively, these findings position TUDCA as a pleiotropic modulator of ER stress, mitochondrial integrity, inflammation, and insulin signalling [3,5,6].
Clinically, TUDCA has been most extensively evaluated in hepatology and neurology, with growing, though still preliminary data in metabolic disease. In cholestatic and other chronic liver diseases, TUDCA and related bile acids have been used as alternative or adjunct therapies to improve cholestasis, liver enzymes, and histological markers, supported by its hepatocellular cytoprotective and anti‑apoptotic effects. Early‑phase metabolic studies, including small trials and mechanistic protocols, suggest that TUDCA can improve insulin sensitivity and liver biochemistry in conditions such as NAFLD or protease‑inhibitor–associated insulin resistance, although sample sizes are modest and follow‑up is relatively short [3].
In neurodegenerative disease, phase II studies and an ongoing phase III trial in amyotrophic lateral sclerosis (ALS) have explored TUDCA as a disease‑modifying adjunct based on its ER‑stress and mitochondria-targeted actions. Pilot data indicate that TUDCA, often combined with riluzole, is safe over 12–18 months and may slow functional decline, prompting larger confirmatory trials; patents and exploratory work also extend to Huntington’s, Parkinson’s, and Alzheimer’s disease where similar pathogenic pathways operate. However, despite robust preclinical literature and encouraging early human studies across hepatology, metabolism, and neurology, long‑term cardiometabolic outcome data, such as effects on major cardiovascular events, hard liver endpoints, or all-cause mortality remain lacking, underscoring the need for larger, adequately powered randomized trials before TUDCA can be fully positioned within cardiometabolic and aging-medicine paradigms [3,5,9,10].
Rationale For Synergy: Cardiometabolic and Aging Context
Cardiometabolic diseases and biological aging share a constellation of converging pathophysiological mechanisms that operate across molecular, cellular, and systems levels, creating a feed‑forward cycle of vascular and metabolic deterioration. Central among these mechanisms is chronic low‑grade inflammation, often termed “inflammaging,” which is characterized by persistent elevation of pro‑inflammatory cytokines such as interleukin‑6 (IL‑6), tumor necrosis factor‑α (TNF‑α), and C‑reactive protein (CRP), alongside oxidative stress resulting from excessive reactive oxygen species (ROS) generation and inadequate antioxidant defense. This inflammatory and oxidative milieu damages endothelial cells, promotes eNOS uncoupling, thereby reducing nitric oxide bioavailability and activates nuclear factor- κB (NF‑κB) signalling cascades that perpetuate endothelial dysfunction, leukocyte adhesion, and smooth muscle cell proliferation, ultimately accelerating atherosclerosis, arterial stiffness, and end‑organ damage [11,12,13].

A second hallmark of both cardiometabolic disease and aging is hypercoagulability and endothelial dysfunction. Aging and metabolic disorders such as diabetes and obesity increase circulating fibrinogen, plasminogen activator inhibitor‑1 (PAI‑1), and D‑dimer, shifting the hemostatic balance toward a prothrombotic state that promotes intravascular thrombus formation and impairs microcirculatory perfusion. Concurrently, endothelial dysfunction marked by reduced flow-mediated dilation, increased endothelial microparticle shedding, and upregulation of adhesion molecules and procoagulant factors disrupts the normally anti-inflammatory endothelial phenotype, contributing to atherosclerotic plaque progression, instability, and thrombotic occlusion. Endothelial dysfunction has emerged as one of the earliest markers of vascular aging and a bidirectional driver of cardiometabolic disease, as metabolic perturbations induce endothelial injury while endothelial impairment exacerbates insulin resistance, hypertension, and atherogenesis [11,12,13,14,15,16,17].
At the subcellular level, ER stress, mitochondrial dysfunction, and impaired autophagy represent critical nodes linking aging and cardiometabolic pathology. During aging, the accumulation of misfolded and aggregated proteins overwhelms the capacity of ER quality control systems, triggering the unfolded protein response (UPR) and chronic ER stress in cardiomyocytes, endothelial cells, and hepatocytes. ER stress, in turn, is tightly coupled to mitochondrial dysfunction: it impairs mitochondria‑ER contact sites (MAMs), disrupts calcium homeostasis, reduces ATP production via oxidative phosphorylation, and increases mitochondrial ROS leakage, thereby initiating apoptosis and cellular senescence. In parallel, age‑related decline in autophagy, particularly mitophagy result in inefficient clearance of damaged mitochondria and protein aggregates, perpetuating oxidative injury and ER stress in a vicious cycle. This triad of ER stress, mitochondrial dysfunction, and defective autophagy is now recognized as a unifying pathogenic mechanism in cardiac aging, heart failure with preserved ejection fraction, atherosclerosis, and diabetic cardiomyopathy [18,19,20,21,22,23].
Within this interconnected network of aging and cardiometabolic pathophysiology, nattokinase and TUDCA occupy complementary mechanistic niches. Nattokinase primarily targets the thrombotic and hemodynamic axis: its direct fibrinolytic activity and capacity to reduce fibrinogen burden, blood viscosity, and platelet aggregation position it as an intervention against the hypercoaguable state and microvascular dysfunction that characterize both metabolic syndrome and vascular aging. by improving blood flow, reducing thrombus formation, and potentially stabilizing atherosclerotic plaques through modulation of lipid and inflammatory parameters, nattokinase addresses downstream hemostatic and vascular consequences of inflammaging and endothelial injury [1,2,3,6].
Conversely, TUDCA acts predominantly on the cellular stress and metabolic axis: its capacity to mitigate ER stress, preserve mitochondrial integrity, reduce oxidative damage, and improve insulin sensitivity targets upstream molecular drivers of cardiometabolic disease and aging. by alleviating ER-stress-mediated apoptosis and inflammation in endothelial cells, hepatocytes, and cardiomyocytes, enhancing mitochondrial membrane potential, and restoring proteostasis and autophagic flux, TUDCA addresses fundamental cellular dysfunction that precedes and exacerbates vascular and metabolic phenotypes. Its effects on bile-acid signalling and hepatic lipid metabolism further complement systemic metabolic homeostasis, reducing lipotoxicity and atherogenic dyslipidemia [3,5,6,18,21].
Taken together, nattokinase and TUDCA represent biologically rational pairing that engages the converging pathways of aging and cardiometabolic disease from orthogonal but synergistic angles: nattokinase mitigates thrombo-inflammatory burden and hemodynamic dysfunction, while TUDCA restores cellular resilience at the level of ER and mitochondrial homeostasis. This dual-axis approach addresses both the macrovascular (thrombosis, atherosclerosis, arterial stiffness) and microvascular /cellular (ER stress, mitochondrial decay, metabolic dysregulation) dimensions of cardiometabolic aging, providing a mechanistic foundation for exploring their combined use in metabolic and vascular wellness strategies [1,2,3,5,6,11,18].
Complementary Mechanisms
This complementary mechanisms of nattokinase and TUDCA span three interconnected domains which are vascular and hemorheologic, metabolic and hepatic, and cellular stress and aging, creating a multi-dimensional framework for cardiometabolic and longevity interventions [1,2,24].
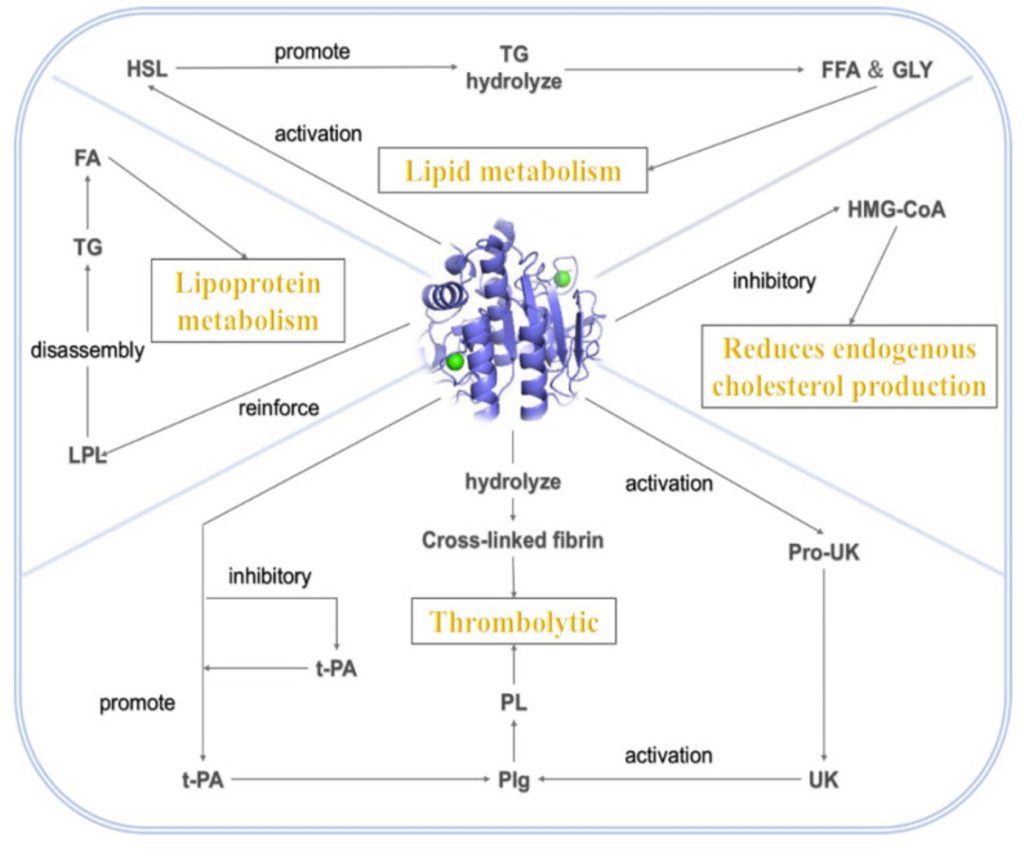
In the vascular and hemorheologic domain, nattokinase exerts pleiotropic effects that extend beyond simple fibrinolysis to encompass comprehensive modulation of thrombotic risk and microvascular function. Through direct fibrin degradation and indirect activation of endogenous fibrinolytic pathways, nattokinase reduces fibrinogen burden, improves clot resolution, and decreases whole-blood viscosity, thereby lowering the risk of both macrovascular thrombotic events such as myocardial infarction and ischemic stroke and microvascular perfusion deficits that contribute to end-organ damage. Meta-analyses of randomized controlled trials confirm that nattokinase supplementation over extended periods (typically 26 weeks or longer) significantly reduces systolic and diastolic blood pressure in prehypertensive and mildly hypertensive populations, with effects mediated in part through improved hemorheology and reduced vascular resistance. Complementing these hemodynamic benefits, TUDCA acts on the endothelium through distinct cellular pathways: preclinical models demonstrate that TUDCA administration prevents hyperglycemia-induced endothelial dysfunction by reducing ER stress and oxidative damage, and directly stimulates angiogenic activity in human vascular endothelial cells via activation of the Akt/eNOS pathway, enhancing endothelial nitric oxide synthase (eNOS) phosphorylation and nitric oxide (NO) bioavailability. This upregulation of NO signaling supports endothelium-dependent vasodilation, inhibits platelet aggregation, and protects against oxidative stress, effects that are mechanistically orthogonal to but functionally synergistic with nattokinase’s hemorheologic improvements. Taken together, nattokinase and TUDCA address vascular health from complementary angles: nattokinase by reducing thrombotic burden and improving blood flow dynamic, and TUDCA by restoring endothelial homeostasis and NO-mediated vascular protection [1,2,24,25,26,27,28,29].

In the metabolic and hepatic domain, TUDCA and nattokinase similarly exert convergent effects on atherogenic lipid metabolism and hepatic function through distinct mechanisms. TUDCA’s metabolic benefits are well-documented in animal models of obesity and non-alcoholic fatty liver disease (NAFLD), where oral administration improves insulin signaling via attenuation of ER-stress-mediated impairment of insulin receptor substrate-1 (IRS-1) function, reduces hepatic steatosis through enhanced fatty acid oxidation and decreased lipogenesis, and mitigates lipotoxic ER stress that drives hepatocyte apoptosis and progression to non-alcoholic steatohepatitis (NASH). hese hepatic actions translate into systemic metabolic improvements, including reductions in fasting glucose, HOMA-IR, and circulating inflammatory cytokines, thereby addressing upstream drivers of atherogenic dyslipidemia characteristic of metabolic syndrome. Nattokinase, meanwhile, acts on the lipid profile through multiple pathways: it activates hormone-sensitive lipase (HSL) in adipocytes to promote triglyceride hydrolysis and lipolysis, enhances lipoprotein lipase (LPL) activity to accelerate triglyceride-rich lipoprotein clearance, and inhibits hepatic HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis. Large-scale clinical trials and meta-analyses confirm that nattokinase (at doses ranging from 2,000 to 10,800 FU/day for 12 to 26 weeks) significantly reduces total cholesterol by 10–15.9%, triglycerides by 12–15.7%, and LDL cholesterol by 11–18.1%, while increasing HDL cholesterol by 8–15.8%, with corresponding reductions in carotid intima-media thickness (21.7% reduction) and atherosclerotic plaque area (36% reduction). By improving blood rheology and reducing viscosity-related vascular shear stress, nattokinase may also act upstream on atherosclerotic plaque initiation and progression, complementing TUDCA’s hepatic lipid-lowering and insulin-sensitizing effects and creating a synergistic metabolic intervention that addresses both lipid synthesis/ clearance and systemic inflammatory and thrombotic drivers of atherosclerosis [2,3,5,27,28,29,30].
In the cellular stress and aging domain, TUDCA and nattokinase address fundamental biological mechanisms that underlie both cardiometabolic disease and the aging process itself. TUDCA’s capacity to attenuate ER stress and apoptosis directly intersects with pathways implicated in cellular senescence and organ aging: chronic ER stress, which accumulates with age due to declining UPR capacity and rising proteotoxic burden, activates pro-senescence signalling via the ATF4-CHOP pathway and triggers apoptosis through PERK, IRE1α, and ATF6-mediated caspase activation. By reducing CHOP expression, preserving protein-folding homeostasis, and preventing mitochondrial-mediated apoptosis, TUDCA limits the accumulation of senescent cells and maintains tissue regenerative capacity, effects demonstrated in preclinical models of cardiac aging, lung senescence, and hepatic steatosis. Furthermore, TUDCA’s preservation of mitochondrial membrane potential and reduction in mitochondrial ROS generation mitigate oxidative damage to nuclear and mitochondrial DNA, lipids, and proteins, slowing fundamental aging processes such as telomere attrition and epigenetic drift. Complementing these cellular protective effects, nattokinase’s reduction of thrombo-inflammatory load may lower cumulative ischemia-reperfusion injury and microvascular damage that accumulate with age and accelerate end-organ decline. By improving microcirculatory perfusion and reducing micro-thrombus formation, nattokinase supports nutrient and oxygen delivery to tissues, facilitates waste removal, and reduces hypoxic stress that triggers senescence and fibrosis in cardiac, renal and cerebral tissues. Additionally, nattokinase’s anti-inflammatory and antioxidant properties, evidenced by reductions in serum inflammatory markers in clinical cohorts may attenuate inflammaging and oxidative stress that drive cellular senescence and age-related dysfunction. Thus, TUDCA and nattokinase together target both the intercellular stress responses (ER stress, mitochondrial dysfunction, apoptosis) and extracellular/systemic stresses (thrombosis, ischemia, inflammation) that constitute the mechanistic core of cardiometabolic aging, offering a biologically rational dual-axis strategy for metabolic and vascular wellness across the lifespan [1,2,3,6,18,21,27,29,31,32].
Hypothesized Benefits of Combination
The mechanistic convergence of nattokinase and TUDCA suggests several hypothetical synergistic outcomes that span vulnerable plaque stabilization, cardiometabolic flexibility and systemic vascular-metabolic integration, although it must be emphasized that these remain mechanistic extrapolations in the absence of direct clinical trials evaluating the combination [1,2,3,5].
Enhanced protection of vulnerable atherosclerotic plaques represents a particularly compelling theoretical benefit of combined nattokinase and TUDCA therapy. Vulnerable plaques characterized by thin fibrous caps, large necrotic cores, abundant macrophages, and reduced smooth muscle cell content are prone to rupture, leading to acute thrombotic occlusion and clinical events such as myocardial infarction and stroke. The structural integrity of the fibrous cap depends critically on vascular smooth muscle cells (VSMCs), which synthesize collagen types I and III; consequently, VSMC apoptosis within plaques weakens the cap, enlarges the necrotic core, and predisposes to rupture, with increased VSMC apoptosis consistently observed in ruptured versus stable lesions. TUDCA’s capacity to reduce ER stress and apoptosis in vascular cells is directly relevant to this pathology: by attenuating UPR-mediated CHOP expression and preserving mitochondrial membrane potential in endothelial cells and VSMCs, TUDCA could reduce apoptotic cell loss in the fibrous cap, maintain collagen synthesis, and promote plaque stability. Complementing this cellular protection, nattokinase addresses the thrombotic component of plaque vulnerability by reducing fibrin burden within and atop atherosclerotic lesions. Fibrin is a major structural component of thrombi formed on plaque surfaces and accumulates within plaques as a result of local coagulation activation, contributing to plaque growth, inflammation, and the transition from stable to vulnerable morphology. Intraplaque fibrin is more common in symptomatic than asymptomatic lesions, and fibrin degradation products are associated with larger necrotic cores and subclinical plaque rupture or erosion. Nattokinase’s fibrinolytic activity both direct fibrin degradation and enhancement of endogenous plasminogen activation could reduce intraplaque and surface fibrin accumulation, mitigate thrombus-driven plaque expansion, and lower the risk of occlusive thrombosis following plaque rupture. Furthermore, by improving blood rheology and reducing shear stress in stenotic regions, nattokinase may indirectly protect VSMCs from high-shear-stress-induced apoptosis, which is a recognized mechanism of vulnerable plaque formation in high-stress arterial segments. Thus, the combination of nattokinase (reducing fibrin and thrombus formation, improving hemorheology) and TUDCA (reducing ER stress and apoptosis in endothelial and smooth muscle cells) offers a dual-mechanism approach to plaque stabilization that targets both cellular integrity and thrombotic risk in a coordinated manner [1,2,6,24,33,34,35,36,37,38,39].
Improved cardiometabolic flexibility constitutes a second major hypothesized benefit of the nattokinase-TUDCA combination. Metabolic flexibility , defined as the capacity to adapt fuel oxidation to fuel availability, switching efficiently between lipid and glucose utilization in response to fasting, feeding and exercise is impaired in insulin-resistant and cardiometabolic disease states, contributing to intramyocellular lipid accumulation, mitochondrial dysfunction, and progression of metabolic syndrome. Metabolic inflexibility occurs early in cardiometabolic disease and is closely linked to insulin resistance, elevated fasting respiratory quotient, impaired glucose disposal, and reduced capacity for tissue-level substrate switching. The nattokinase-TUDCA combination could address multiple determinants of metabolic inflexibility simultaneously: TUDCA improves insulin signalling via attenuation of ER-stress-mediated inhibition of insulin receptor substrate pathways, reduces hepatic steatosis and lipotoxicity, and enhances mitochondrial function, effects that restore cellular substrate oxidation capacity and normalize fuel selection in liver, skeletal muscle, and adipose tissue. Concurrently, nattokinase targets blood flow and liver-derived metabolic risk factors by improving microcirculatory perfusion (thereby enhancing nutrient and insulin delivery to metabolic tissues), reducing plasma fibrinogen (an inflammatory acute-phase reactant elevated in metabolic syndrome), and modulating lipid metabolism to lower atherogenic dyslipidemia. By enhancing tissue perfusion and reducing blood viscosity, nattokinase may also mitigate hypoxic stress and oxidative damage in insulin-sensitive tissues, which are known to impair mitochondrial oxidative capacity and substrate switching. Additionally, both agents attenuate systemic inflammation, a core driver of insulin resistance and metaboli inflexibility through distinct mechanisms: TUDCA via reduction of ER-stress-induced inflammatory cytokine production and nattokinase via modulation of platelet-leukocyte interactions and inflammatory marker levels. This multi-level intervention simultaneously targeting insulin signalling and ER stress (TUDCA), lipid metabolism and hemorheology (nattokinase), and systemic inflammation (both) offers a comprehensive strategy to restore metabolic flexibility, improve glucose and lipid handling and reduce cardiometabolic risk factors in individuals with metabolic syndrome, prediabetes, or early type 2 diabetes [1,2,3,5,6,27,28,29,40,41,42,43].
It must be emphasized, however, that these proposed synergistic outcomes are mechanistic extrapolations derived from separate preclinical and clinical evidence for nattokinase and TUDCA as monotherapies; no direct clinical trials, preclinical studies, or even case series have formally evaluated the safety, pharmacokinetics, pharmacodynamics, or efficacy of nattokinase plus TUDCA in combination. The theoretical synergy is predicated on the assumption that the two agents will engage their respective molecular targets (fibrinolytic/hemorheologic pathways for nattokinase; ER stress/mitochondrial/metabolic pathways for TUDCA) without antagonism, excessive off-target effects, or unforeseen pharmacological interactions. While the mechanistic orthogonality and complementary biological actions strongly support the plausibility of synergy, rigorous validation through controlled experimental studies, beginning with in vitro co-treatment assays, progressing to animal models of atherosclerosis and metabolic disease, and ultimately advancing to phase I/II human trials is essential before clinical recommendations can be made. Such trials should evaluate not only efficacy endpoints (plaque characteristics, metabolic biomarkers, vascular function) but also safety parameters, particularly given nattokinase’s fibrinolytic activity and TUDCA’s effects on bile acid homeostasis, to ensure that the combination does not introduce unexpected bleeding risk, hepatic dysfunction, or other adverse outcomes. until such evidence is available, the nattokinase-TUDCA combination should be regarded as a hypothesis-generating, biologically rational strategy warranting systematic investigation rather than an evidence-based therapeutic recommendations [1,2,3,5,6].
Potential Applications in Metabolic and Aging Wellness
The hypothesized synergistic effects of combined nattokinase and TUDCA therapy suggest potential applications across several high-risk populations characterized by residual cardiometabolic risk despite conventional management, positioning the combination as a biologically targeted adjunctive strategy rather than a standalone or first-line intervention [1,2,3,44,45].
Individuals with metabolic syndrome, prediabetes, or insulin resistance accompanied by early vascular changes represent a primary target population for nattokinase-TUDCA combination therapy. Metabolic syndrome affects an estimated 25–35% of adults in developed nations and confers a two- to three-fold increased risk of atherosclerotic cardiovascular disease and type 2 diabetes, driven by insulin resistance, atherogenic dyslipidemia (low HDL-C, elevated triglycerides), hypertension, prothrombotic state, and systemic inflammation. Even when treated with statins to guideline-recommended LDL-C targets, patients with metabolic syndrome exhibit substantial residual cardiovascular risk: major statin trials consistently document event rates of 20–30% over follow-up periods of three to seven years despite intensive statin therapy achieving LDL-C levels below 70 mg/dL, with diabetic and metabolic syndrome subgroups showing particularly high residual risk. This persistent risk is attributable to metabolic abnormalities inadequately addressed by LDL-lowering alone, including elevated triglycerides, low HDL-C, insulin resistance, ER stress, oxidative stress, hypercoagulability, and endothelial dysfunction. For such individuals, the nattokinase-TUDCA combination offers orthogonal mechanisms: TUDCA targeting insulin resistance, hepatic steatosis, and ER-stress-mediated inflammation, while nattokinase addresses hypercoagulability, blood viscosity, and lipid abnormalities, potentially filling therapeutic gaps left by statin monotherapy. Subclinical atherosclerosis markers including elevated carotid intima-media thickness, coronary artery calcium scores above the 75th percentile, or endothelial dysfunction on flow-mediated dilation testing further identify metabolic syndrome patients who may benefit from intensified adjunctive intervention [1,2,3,5,27,44,45,46,47,48,49].
Patients with non-alcoholic fatty liver disease (NAFLD) or non-alcoholic steatohepatitis (NASH) and elevated cardiometabolic risk constitute a second high-priority population. NAFLD affects approximately 25% of the global adult population and is strongly associated with obesity, insulin resistance, type 2 diabetes, and metabolic syndrome; cardiovascular disease, not liver failure, is the leading cause of mortality in NAFLD patients, accounting for up to 40% of deaths. Meta-analyses consistently demonstrate that NAFLD confers a 37–69% increased risk of incident cardiovascular disease and a 2.3-fold increased risk of coronary heart disease independent of traditional risk factors, with patients exhibiting NASH or advanced fibrosis (F3–F4 stage) at particularly high risk for major adverse cardiovascular events, cardiac arrhythmias (especially atrial fibrillation), and cardiovascular mortality. The pathophysiological link between NAFLD and cardiovascular disease involves shared drivers such as insulin resistance, atherogenic dyslipidemia, oxidative stress, and chronic inflammation, as well as liver-specific contributions including hepatic secretion of pro-atherogenic lipoproteins, pro-inflammatory cytokines, and procoagulant factors. NAFLD patients also exhibit higher rates of arterial hypertension (16% increased risk), subclinical atherosclerosis (significantly increased carotid intima-media thickness and coronary artery calcification), and ventricular arrhythmias. In this population, TUDCA’s established hepatoprotective effects which are reducing hepatic steatosis, improving insulin sensitivity, attenuating ER stress and lowering liver enzymes in animal models and early human trials address primary hepatic pathology, while nattokinase’s lipid lowering, fibrinolytic, and anti-inflammatory actions target downstream cardiovascular srrquelae. The combination thus offers a dual hepatocardiovascular intervention particularly suited to NAFLD patients with concomitant metabolic syndrome, type 2 diabetes, or evidence of subclinical atherosclerosis [1,2,3,5,27,50,51,52,53,54,55].
Aging individuals with evidence of increased arterial stiffness, hypercoagulability, or subclinical atherosclerosis who remain incompletely optimized on standard therapies represent a third target population. Arterial stiffness, characterized by loss of aortic elasticity, elevated pulse wave velocity, and increased systolic blood pressure is one of the earliest markers of vascular aging and an independent predictor of cardiovascular events, heart failure, stroke, cognitive decline, and all-cause mortality even after adjustment for traditional risk factors. arotid-femoral pulse wave velocity increases from approximately 5.5 m/s at age 10 years to over 11 m/s by age 80, with a particularly steep rise after age 55, and is closely associated with endothelial dysfunction, oxidative stress, vascular smooth muscle cell senescence, and extracellular matrix remodeling. Despite guideline-directed statin and antihypertensive therapy, long-term follow-up studies document cumulative cardiovascular event rates of 15–40% over five to ten years in older adults with established cardiovascular disease, with residual risk persisting even when LDL-C is reduced to 55–70 mg/dL and blood pressure is controlled. Subclinical atherosclerosis, detected by coronary artery calcium scoring, carotid intima-media thickness measurement, or ankle-brachial index identifies individuals at intermediate or high risk who may benefit from more intensive or multi-targeted preventive strategies beyond statin and aspirin monotherapy. For this aging population, the nattokinase-TUDCA combination addresse fundamental mechanisms of vascular aging: nattokinase’s hemorheologic, and fibrinolytic effects may improve microcirculatory perfusion and reduce arterial stiffness-related thrombotic risk, while TUDCA’s attenuation of ER stress, mitochondrial dysfunction, and oxidative damage targets cellular aging processes in vascular smooth muscle cells and endothelium. Current treatment guidelines acknowledge that non-pharmacological interventions (aerobic exercise, dietary sodium restriction) and select antihypertensives can modestly reduce arterial stiffness, but novel therapeutic targets remain under-explored; nutraceutical adjuncts with mechanistic rationale represent a reasonable avenue for investigation in this population [1,2,3,6,12,24,25,45,47,48,49,56].
Integration of nattokinase and TUDCA with conventional cardiometabolic therapy should be conceptualized as adjunctive rather than substitutive, complementing, not replacing, evidence-based pharmacotherapy and lifestyle modification. For individuals on stable statin, antihypertensive, and antiplatelet regimens who continue to exhibit metabolic dysregulation (elevated HOMA-IR, persistent hypertriglyceridemia despite statin therapy, hepatic steatosis), biomarkers of thrombotic risk (elevated fibrinogen, D-dimer), or subclinical atherosclerosis progression, the combination offers a mechanistically targeted add-on strategy that engages pathways orthogonal to LDL-lowering and blood pressure control. This adjunctive positioning is particularly relevant given that lifestyle modification, including dietary intervention, weight loss, and physical activity remains the cornerstone of residual risk management in metabolic syndrome and NAFLD, with pharmacological and nutraceutical interventions serving to enhance rather than circumvent lifestyle efforts [1,2,3,5,44,45,47,48,52].
Specific therapeutic niches where nattokinase-TUDCA combination may offer unique value include patients intolerant to or inadequately responsive to conventional agents. For example, individuals who discontinue statin therapy due to myalgias, hepatotoxicity, or other adverse effects but remain at elevated cardiovascular risk may benefit from nattokinase’s lipid-lowering and anti-atherosclerotic actions as a partial pharmacological gap-filler, while TUDCA addresses metabolic and hepatic dysfunction without the myopathic risk profile of statins. Similarly, patients with contraindications to antiplatelet therapy (due to bleeding risk) but evidence of hypercoagulability might be candidates for nattokinase’s fibrinolytic effects under careful monitoring, though caution regarding bleeding risk remains paramount. Individuals seeking evidence-informed adjunctive support beyond standard therapy, particularly those engaged in metabolic wellness and longevity medicine programs represent another niche, provided such use occurs under medical supervision with systematic monitoring of coagulation parameters, liver function, metabolic biomarkers, and vascular surrogate endpoints [1,2,3,5,27,44,45].
Importantly, all proposed applications remain investigational pending rigorous preclinical and clinical validation of the nattokinase-TUDCA combination, and clinicians considering such use should implement robust safety monitoring protocols, including baseline and serial assessments of coagulation indices (PT/INR, aPTT, fibrinogen, D-dimer), liver enzymes, lipid panels, inflammatory markers, and vascular function testing to evaluate both efficacy signals and adverse effects [1,2,3,44,45].
Safety Considerations and Limitations
The safety profile and limitations of nattokinase and TUDCA, individually and in combination, warrant careful consideration before deployment as adjunctive therapies in metabolic and aging wellness programs. For nattokinase, the principal safety concern relates to its fibrinolytic and antithrombotic properties, which may potentiate bleeding risk, particularly when co‑administered with anticoagulants (e.g., warfarin, DOACs) or antiplatelet agents (e.g., aspirin, clopidogrel). Although clinical trials and observational studies generally report good tolerability with few major hemorrhagic events, the mechanistic potential for increased bleeding through enhanced fibrin degradation and modulation of coagulation factors, renders nattokinase elatively contraindicated in patients with active bleeding, recent major surgery or trauma, hemorrhagic stroke, known bleeding disorders, or severe thrombocytopenia, and it should be used cautiously in those on multi‑agent antithrombotic regimens. An additional limitation is the considerable variability in commercial nattokinase preparations: products differ in fibrinolytic unit (FU) standardization, manufacturing processes, and impurity profiles, and independent analyses have documented discrepancies between labeled and actual activity content, underscoring the need for pharmaceutical‑grade standardization and third‑party quality control if nattokinase is to be integrated into evidence‑based clinical practice [1,2,3,5,27,29,30].
In contrast, TUDCA is generally regarded as well tolerated, with a safety profile largely derived from its use in cholestatic liver disease and early‑phase metabolic and neurodegenerative trials. Reported adverse effects are typically mild and dose‑related, most commonly gastrointestinal symptoms such as diarrhea, abdominal discomfort, and, less frequently, nausea, which tend to emerge at higher daily doses and often improve with dose reduction or administration with food. Serious hepatotoxicity has not been a prominent signal in clinical studies, consistent with TUDCA’s hepatoprotective biology, although elevations in liver enzymes should still be monitored, especially in patients with pre‑existing liver disease or polypharmacy. A key limitation is that long‑term safety data in otherwise healthy or primary‑prevention populations are sparse: most human data come from patients with cholestatic liver disease, NAFLD, or ALS enrolled in relatively small trials of limited duration (typically 6–24 months), leaving substantial uncertainty about the effects of chronic, multi‑year TUDCA supplementation on bile‑acid homeostasis, gallstone risk, microbiome composition, and cardiometabolic endpoints. Consequently, extrapolating short‑term safety in diseased cohorts to long‑term use in generally healthy individuals should be approached with caution [3,4,5].
For the nattokinase–TUDCA combination specifically, safety data are essentially absent: no systematic preclinical toxicology, pharmacokinetic interaction studies, or human trials have evaluated co‑administration, leaving potential additive, synergistic, or antagonistic safety effects unknown. Theoretically, combining a fibrinolytic enzyme with a bile‑acid–based cytoprotective agent raises several considerations. First, polypharmacy is common in the target populations (metabolic syndrome, NAFLD, older adults with subclinical atherosclerosis), and interactions with anticoagulants, antiplatelets, antihypertensives, and glucose‑lowering drugs may alter bleeding risk, blood pressure, glycemic control, or hepatic metabolism; thus, comprehensive medication review and monitoring are essential. Second, hepatic and renal function should be carefully assessed, as TUDCA’s pharmacokinetics depend on hepatic uptake and enterohepatic circulation, while impaired renal function may influence the clearance of fibrin degradation products and other metabolites relevant to nattokinase’s activity and safety. Third, baseline coagulation status including platelet count, PT/INR, aPTT, fibrinogen, and D-dimer should be evaluated and periodically reassessed to detect subclinical bleeding tendency or excessive fibrinolysis, especially in patients receiving concurrent antithrombotic therapy. Until formal dose‑finding and safety studies are conducted, conservative dosing, stepwise titration, and close laboratory and clinical monitoring are prudent if the combination is used in research or highly selected clinical contexts [1,2,3,4,5,29,30,44,45,52].
Regulatory and evidentiary limitations further constrain the clinical positioning of nattokinase and TUDCA in metabolic and aging medicine. In most jurisdictions, both compounds are predominantly regulated and marketed as dietary supplements or nutraceuticals rather than as approved drugs, which means that manufacturing quality, standardization of active content (FU for nattokinase, milligram dose and purity for TUDCA), and contamination control may vary widely between brands and regions. This heterogeneity complicates dose–response extrapolation from clinical studies using specific standardized preparations to real‑world supplement products. From an evidence standpoint, there is a notable lack of large randomized controlled trials with hard cardiovascular or mortality endpoints for either agent: existing nattokinase data are largely limited to small RCTs and open‑label studies assessing surrogate markers such as blood pressure, lipid profile, and carotid intima–media thickness, while TUDCA trials focus on liver enzymes, insulin sensitivity, or functional scales in ALS rather than myocardial infarction, stroke, or all‑cause mortality. For the combination, clinical outcome data are completely absent. As a result, while mechanistic plausibility and early surrogate‑endpoint data support further investigation, current evidence does not justify positioning nattokinase, TUDCA, or their combination as replacements for statins, antihypertensives, antiplatelets, or other guideline‑directed therapies, and their use should remain adjunctive and preferably within research frameworks until robust, adequately powered outcome trials are available [1,2,3,4,5,27,29,30,44,45,56].
Future Research Directions
Future research on the combined use of nattokinase and TUDCA should progress along a translational continuum from mechanistic preclinical models to pragmatic outcome‑oriented clinical trials, with biomarker‑based stratification embedded throughout. Preclinically, dedicated studies in animal models of atherosclerosis and metabolic syndrome are needed to compare combination therapy against each monotherapy and control, focusing on plaque stability (fibrous cap thickness, necrotic core size, intraplaque fibrin, and smooth muscle cell content), insulin sensitivity (hyperinsulinemic–euglycemic clamp, tissue‑specific insulin signalling), and organ‑specific ER stress and mitochondrial dysfunction in liver, vasculature, myocardium, and adipose tissue. Such studies should incorporate detailed pharmacokinetic/pharmacodynamic (PK/PD) profiling, coagulation assays, and histological endpoints to clarify dose ranges that maximize synergistic effects on fibrinolysis and ER‑stress mitigation while minimizing bleeding or bile‑acid‑related toxicity [1,2,3,6,18,51].
Building on robust preclinical data, early‑phase human trials should begin with phase 1 and 2 studies designed primarily around safety, tolerability, and mechanistic readouts rather than clinical events. Phase 1 trials in carefully selected adults with metabolic syndrome, NAFLD, or subclinical atherosclerosis could evaluate graded dosing of nattokinase plus TUDCA, with intensive PK/PD sampling and serial monitoring of coagulation parameters (PT/INR, aPTT, fibrinogen, D‑dimer), endothelial function using flow‑mediated dilation (FMD), and circulating inflammatory markers such as hs‑CRP and IL‑6. Phase 2 studies could then expand to larger cohorts to test predefined dose regimens over 3–12 months, incorporating metabolic endpoints (HOMA‑IR, oral glucose tolerance, fasting lipids), liver enzymes and non‑invasive hepatic fat quantification (MRI‑PDFF) in NAFLD subsets, and vascular surrogate markers including carotid intima–media thickness and pulse wave velocity. These trials would provide critical data on feasibility, short‑term efficacy signals, and composite safety profiles required to justify progression to larger outcomes studies [1,2,3,5,55,56,57,58,59,60,61,62,63,64,65,66,67].
Longer‑term goals should include pragmatic randomized controlled trials in high‑risk metabolic or aging populations that integrate both surrogate imaging endpoints and, ultimately, hard cardiovascular and hepatic outcomes. In intermediate‑sized trials (e.g., 300–1,000 participants), primary endpoints could center on changes in carotid intima–media thickness, progression of coronary artery calcium scores, and liver fat content by MRI‑PDFF, all of which are validated surrogate markers used extensively in atherosclerosis and NAFLD/NASH drug development. With sufficient follow‑up and event accrual, larger pragmatic trials could then examine clinical outcomes such as major adverse cardiovascular events (MI, stroke, cardiovascular death), incident heart failure, need for coronary or peripheral revascularization, and, in NAFLD cohorts, progression to advanced fibrosis or cirrhosis. Across all stages, a biomarker‑driven personalization strategy should be incorporated: baseline levels of fibrinogen and D‑dimer (as indices of thrombotic risk), hs‑CRP and IL‑6 (systemic inflammation), HOMA‑IR and fasting triglycerides (insulin resistance and metabolic status), liver stiffness and MRI‑PDFF (hepatic disease burden), and ER‑stress‑related markers where available could be used both for enrichment (selecting high‑risk, likely responsive subgroups) and for exploratory responder analyses. Such a precision‑medicine framework would help identify phenotypes in which nattokinase‑TUDCA synergy is most clinically meaningful and inform rational integration of this combination into future cardiometabolic and aging‑medicine care pathways [1,2,3,5,44,45,52,55,56,61,63,64,65].
Conclusion
Nattokinase and TUDCA can be positioned as complementary agents acting on distinct yet convergent axes that underpin cardiometabolic disease and biological aging: nattokinase primarily targets thrombosis, blood rheology and vascular function through its fibrinolytic and antithrombotic effects, whereas TUDCA modulates ER-stress driven cellular dysfunction, mitochondrial integrity, and metabolic signaling in key organs such as the liver, vasculture, and myocardium. Together, these mechanisms intersect at critical nodes of endothelial dysfunction, atherogenesis, insulin resistance, and organ-level resilience, providing a coherent biological rationale for their combined use in cardiometabolic and aging-wellness frameworks.
Within this context, the combined use of nattokinase and TUDCA emerges as a biologically plausible and potentially synergistic strategy, insofar as it simultaneously addresses macro- and microvascular thrombotic burden, blood pressure and hemorheology, hepatic steatosis and insulin resistance, and ER-stress-mediated cellular injury. Conceptually, such a dual-axis approach could enhance plaque stability, improve metabolic flexibility, and attenuate inflammaging more effectively than either agent alone. However, it is crucial to recognize that the current evidence base for this synergy is indirect and largely mechanistic, derived from separate preclinical and clinical studies each compound rather than from dedicated evaluations of the combination.
In light of these limitations, nattokinase plus TUDCA should presently be regarded as an experimental, adjunctive approach rather than an established therapeutic option. Its use is best confined to research settings or, where permitted, to individualized off-label application under expert supervision, with careful attention to coagulation status, hepatic function, comorbidities, and concomitant pharmacotherapy, especially in patients receiving anticoagulants, antiplatelets, or multiple cardiometabolic agents. It should not be positioned as a substitute for guideline-directed therapies such as statins, antihypertensives, glucose-lowering drugs, or antithrombotic agents, but rather as a potential supplement to a foundation of lifestyle optimization and evidence-based medical care.
To move from hypothesis to clinical utility, well-designed preclinical and clinical studies are urgently needed. Priority areas include mechanistic animal work directly testing combination versus monotherapy effects on plaque biology, insulin sensitivity, ER stress, and organ function; early-phase human trials characterizing safety, pharmacokinetics/pharmacodynamics, and biomarker responses and ultimately, randomized controlled trials in carefully phenotyped high-risk metabolic or aging populations. Such trials should aim to define optimal dosing strategies, delineate short- and long-term safety profiles (including bleeding and hepatobiliary outcomes), clarify additive or synergistic clinical benefits over standard care, and identify biomarker-defined subgroups mostl likely to benefit. Only through this structured translational pathway can the true therapeutic potential and limitations of the nattokinase-TUDCA pairing be established for cardiometabolic and aging wellness.
Reference
- Pham PT, Han B, Hoang BX. Nattospes as Effective and Safe Functional Supplements in Management of Stroke. Journal of Medicinal Food. 2020 Aug 1;23(8):879–85.
- Norwitz N. Nattokinase: The Heart Health Supplement on Everyone’s Tongue [Internet]. Substack.com. StayCurious Metabolism; 2025 [cited 2026 Jan 18]. Available from: https://staycuriousmetabolism.substack.com/p/nattokinase-the-heart-health-supplement
- Wang W, Zhao J, Gui W, Sun D, Dai H, Xiao L, et al. Tauroursodeoxycholic acid inhibits intestinal inflammation and barrier disruption in mice with non-alcoholic fatty liver disease. British Journal of Pharmacology [Internet]. 2018 Feb 1 [cited 2022 Apr 21];175(3):469–84. Available from: https://pubmed.ncbi.nlm.nih.gov/29139555/
- Albanese A, Ludolph AC, McDermott CJ, Corcia P, Van Damme P, Van den Berg LH, et al. Tauroursodeoxycholic acid in patients with amyotrophic lateral sclerosis: The TUDCA-ALS trial protocol. Frontiers in Neurology. 2022 Sep 27;13.
- Israelle Netto Freitas, Alves J, Moreno K, Bruna Lourençoni Alves, Dos T, João Paulo Camporez, et al. Insights by which TUDCA is a potential therapy against adiposity. 2023 Feb 21;14. Available from: https://doi.org/10.3389%2Ffendo.2023.1090039
- Yoon YM, Lee JH, Yun SP, Han YS, Yun CW, Lee HJ, et al. Tauroursodeoxycholic acid reduces ER stress by regulating of Akt-dependent cellular prion protein. Scientific Reports. 2016 Dec;6(1).
- Malo A, B. Krüger, E. Seyhun, C. Schäfer, Hoffmann RT, B. Göke, et al. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, trypsin activation, and acinar cell apoptosis while increasing secretion in rat pancreatic acini. AJP Gastrointestinal and Liver Physiology. 2010 Jul 30;299(4):G877–86.
- Huang X, Wu L, Kuang Y, Li X, Deng X, Liang X, et al. Tauroursodeoxycholic acid mediates endoplasmic reticulum stress and autophagy in adrenocortical carcinoma cells. Oncology Letters. 2019 Nov 5;18(6):6475–82.
- Albanese A, Ludolph AC, McDermott CJ, Corcia P, Van Damme P, Van den Berg LH, et al. Tauroursodeoxycholic acid in patients with amyotrophic lateral sclerosis: The TUDCA-ALS trial protocol. Frontiers in Neurology. 2022 Sep 27;13.
- Keene CD, Rodrigues CMP, Eich T, Chhabra MS, Steer CJ, Low WC. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proceedings of the National Academy of Sciences. 2002 Jul 29;99(16):10671–6.
- Welcome To Zscaler Directory Authentication [Internet]. Nih.gov. 2026 [cited 2026 Jan 18]. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC10932073/
- Herzog MJ, Müller P, Lechner K, Stiebler M, Arndt P, Kunz M, et al. Arterial stiffness and vascular aging: mechanisms, prevention, and therapy. Signal Transduction and Targeted Therapy. 2025 Aug 31;10(1).
- Sun HJ, Wu ZY, Nie XW, Bian JS. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Frontiers in Pharmacology. 2020 Jan 21;10.
- Senst B. Hypercoagulability [Internet]. U.S. National Library of Medicine; 2023 [cited 2026 Jan 18]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538251/
- [Internet]. [cited 2026 Jan 18]. Available from: https://academic.oup.com/eurjpc/advance-article-pdf/doi/10.1093/eurjpc/zwaf673/64703688/zwaf673.pdf
- Zhang J. Biomarkers of endothelial activation and dysfunction in cardiovascular diseases. Reviews in Cardiovascular Medicine. 2022 Feb 22;23(2):073.
- LOEFFEN R, SPRONK HMH, TEN CATE H. The impact of blood coagulability on atherosclerosis and cardiovascular disease. Journal of Thrombosis and Haemostasis. 2012 Jul;10(7):1207–16.
- Ma C, Liu Y, Fu Z. Implications of endoplasmic reticulum stress and autophagy in aging and cardiovascular diseases. Frontiers in Pharmacology. 2024 Jul 25;15.
- Popov LD. Aging and Senescence Associated Mitochondrial Dysfunction: A Target against Cardiovascular Disorders of the Elderly Individuals [Internet]. International Journal of Geriatrics and Gerontology. Gavin Publishers; 2025 [cited 2026 Jan 19]. Available from: https://www.gavinpublishers.com/article/view/aging-and-senescence-associated-mitochondrial–dysfunction-a-target-against-cardiovascular-disorders-of-the-elderly-individuals
- Chen Q, Arun Samidurai, Thompson J, Hu Y, Das A, Willard B, et al. Endoplasmic reticulum stress-mediated mitochondrial dysfunction in aged hearts. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease. 2020 Nov 1;1866(11):165899–9.
- Sakthijothi Muthu, Tran Z, Jayapalraja Thilagavathi, Tanvi Bolarum, Azzam EI, Suzuki CK, et al. Aging triggers mitochondrial, endoplasmic reticulum, and metabolic stress responses in the heart. The Journal of Cardiovascular Aging [Internet]. 2025 Feb 18;5(1). Available from: https://www.oaepublish.com/articles/jca.2024.17
- Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: from mechanisms to therapeutics. American Journal of Physiology-Heart and Circulatory Physiology. 2013 Aug 15;305(4):H459–76.
- Chen C, Dong X, Zhang W, Chang X, Gao W. Dialogue between mitochondria and endoplasmic reticulum-potential therapeutic targets for age-related cardiovascular diseases. Frontiers in Pharmacology. 2024 Jun 13;15:1389202–2.
- Yang SS, Oh JM, Chun S, Kim BS, Kim CS, Lee J. Tauroursodeoxycholic acid induces angiogenic activity in endothelial cells and accelerates bone regeneration. Bone [Internet]. 2019 Oct 15;130:115073. Available from: https://www.sciencedirect.com/science/article/abs/pii/S8756328219303667
- Walsh LK, Restaino RM, Neuringer M, Manrique C, Padilla J. Administration of tauroursodeoxycholic acid prevents endothelial dysfunction caused by an oral glucose load. 2016 Sep 22;130(21):1881–8.
- Lu H, Wu Z, Wan M, Xiong S, Huang X, Liu T, et al. Taurochenodeoxycholic acid alleviates obesity-induced endothelial dysfunction. European Heart Journal [Internet]. 2025 Oct 3 [cited 2026 Jan 19]; Available from: https://academic.oup.com/eurheartj/advance-article/doi/10.1093/eurheartj/ehaf766/8272824?login=false
- Wei C, Cai R, Song Y, Liu X, Xu HL. Research Progress of Nattokinase in Reducing Blood Lipid. Nutrients. 2025 May 24;17(11):1784.
- Li X, Long J, Gao Q, Pan M, Wang J, Yang F, et al. Nattokinase Supplementation and Cardiovascular Risk Factors: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Reviews in Cardiovascular Medicine. 2023 Aug 15;24(8):234–4.
- Maslarov D, Drenska D, Maslarova-Gelov J, Gelov I. Understanding the concept of Nattokinase use: a few years after beginning. Biotechnology & Biotechnological Equipment. 2023 Mar 10;37(1).
- Chen H, McGowan EM, Ren N, Lal S, Nassif N, Shad-Kaneez F, et al. Nattokinase: A Promising Alternative in Prevention and Treatment of Cardiovascular Diseases. Biomarker Insights. 2018 Jan;13:117727191878513.
- Chen X, Shi C, He M, Xiong S, Xia X. Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Signal Transduction and Targeted Therapy [Internet]. 2023 Sep 15;8(1):1–40. Available from: https://www.nature.com/articles/s41392-023-01570-w#Sec24
- Maunick Lefin Koloko Ngassie, Brandsma C, Reinoud Gosens, Prakash YS, Burgess JK. The Stress of Lung Aging: Endoplasmic Reticulum and Senescence Tête-à-Tête. Physiology. 2021 May 1;36(3):150–9.
- Boyle JJ. Vascular smooth muscle cell apoptosis in atherosclerosis. International Journal of Experimental Pathology. 2001 Dec 25;80(4):197–203.
- Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circulation research. 2016 Feb 19;118(4):692–702.
- Walsh LK, Restaino RM, Neuringer M, Manrique C, Padilla J. Administration of tauroursodeoxycholic acid prevents endothelial dysfunction caused by an oral glucose load. 2016 Sep 22;130(21):1881–8.
- Ząbczyk M, Natorska J, Undas A. Fibrin Clot Properties in Atherosclerotic Vascular Disease: From Pathophysiology to Clinical Outcomes. Journal of Clinical Medicine [Internet]. 2021 Jul 5 [cited 2022 Jan 8];10(13):2999. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8268932/
- Hou XZ, Yang YT, Yao JM. Vulnerable plaques in atherosclerosis: focus on angiogenesis-associated phenotypic crosstalk. Frontiers in Pharmacology. 2026 Jan 13;16.
- KAVURMA MM, BHINDI R, LOWE HC, CHESTERMAN C, KHACHIGIAN LM. Vessel wall apoptosis and atherosclerotic plaque instability. Journal of Thrombosis and Haemostasis [Internet]. 2004 Nov 18;3(3):465–72. Available from: https://www.sciencedirect.com/science/article/pii/S1538783622164664
- Mrozinska S, Wypasek E, Elżbieta Broniatowska, Anetta Undas. Accelerated fibrin clot degradation is associated with arterial thromboembolism in patients following venous thrombosis: a cohort study. Scientific Reports [Internet]. 2021 Oct 26 [cited 2026 Jan 19];11(1):21003–3. Available from: https://www.nature.com/articles/s41598-021-00411-6
- Thyfault JP, R. Scott Rector, Noland RC. Metabolic Inflexibility in Skeletal Muscle: A Prelude to the Cardiometabolic Syndrome? Journal of The Cardiometabolic Syndrome. 2006 Jun 1;1(3):184–9.
- Ang JHC, Sun L, Foo SYR, Leow MKS, Vidal-Puig A, Fontana L, et al. Perspectives on whole body and tissue-specific metabolic flexibility and implications in cardiometabolic diseases. Cell Reports Medicine. 2025 Sep;6(9):102354.
- Galgani JE, Moro C, Ravussin E. Metabolic flexibility and insulin resistance. American Journal of Physiology-Endocrinology and Metabolism. 2008 Nov;295(5):E1009–17.
- Malin SK, Haus JM, Solomon TPJ, Blaszczak A, Kashyap SR, Kirwan JP. Insulin sensitivity and metabolic flexibility following exercise training among different obese insulin-resistant phenotypes. American Journal of Physiology-Endocrinology and Metabolism. 2013 Nov 15;305(10):E1292–8.
- Sampson UK, Fazio S, Linton MF. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Current Atherosclerosis Reports [Internet]. 2012 Feb 1;14(1):1–10. Available from: https://pubmed.ncbi.nlm.nih.gov/22102062/
- Vijayaraghavan, Baum S, Desai NR, Voyce SJ. Intermediate and long-term residual cardiovascular risk in patients with established cardiovascular disease treated with statins. Frontiers in cardiovascular medicine. 2024 Jan 15;10.
- Vega GL, Wang J, Grundy SM. Utility of metabolic syndrome as a risk enhancing factor in decision of statin use. Journal of Clinical Lipidology. 2021 Mar;15(2):255–65.
- Mishra SS, Panda A, Patnaik AN. Subclinical Atherosclerosis. Indian Journal of Clinical Cardiology. 2025 Apr 10;6(2):141–7.
- Iurciuc S, Cimpean AM, Mitu F, Heredea R, Iurciuc M. Vascular aging and subclinical atherosclerosis: why such a “never ending” and challenging story in cardiology?. Clinical Interventions in Aging. 2017 Aug;Volume 12:1339–45.
- Andrzej Wykretowicz, Gerstenberger P, Guzik P, Milewska A, Tomasz Krauze, K. Adamska, et al. Arterial stiffness in relation to subclinical atherosclerosis. European Journal of Clinical Investigation. 2009 Jan 1;39(1):11–6.
- Ma W, Wu W, Wen W, Xu F, Han D, Lyu J, et al. Association of NAFLD with cardiovascular disease and all-cause mortality: a large-scale prospective cohort study based on UK Biobank. Therapeutic Advances in Chronic Disease. 2022 Jan 1;13:204062232211224-204062232211224.
- Wójcik-Cichy K, Koślińska-Berkan E, Piekarska A. The influence of NAFLD on the risk of atherosclerosis and cardiovascular diseases. Clinical and Experimental Hepatology. 2018;4(1):1–6.
- Kasper P, Martin A, Lang S, Kütting F, Goeser T, Demir M, et al. NAFLD and cardiovascular diseases: a clinical review. Clinical Research in Cardiology: Official Journal of the German Cardiac Society [Internet]. 2020 Jul 21;110(7). Available from: https://pubmed.ncbi.nlm.nih.gov/32696080/
- Huang DQ, Downes M, Evans RM, Witztum JL, Glass CK, Rohit Loomba. Shared Mechanisms between Cardiovascular Disease and NAFLD. Seminars in liver disease. 2022 Aug 25;42(04):455–64.
- Kevan Josloff, Beiriger J, Khan A, Gawel RJ, Kirby RS, Kendrick AD, et al. Comprehensive Review of Cardiovascular Disease Risk in Nonalcoholic Fatty Liver Disease. Journal of Cardiovascular Development and Disease. 2022 Nov 26;9(12):419–9.
- Li M, Wang H, Zhang X, Cai J, Li H. NAFLD: An Emerging Causal Factor for Cardiovascular Disease. Physiology. 2023 Nov 1;38(6):255–65.
- Cheng DC, Climie RE, Shu M, Grieve SM, Kozor R, Figtree GA. Vascular aging and cardiovascular disease: pathophysiology and measurement in the coronary arteries. Frontiers in Cardiovascular Medicine. 2023 Nov 28;10.
- Frick M, Weidinger F. Endothelial Function: A Surrogate Endpoint in Cardiovascular Studies? Current Pharmaceutical Design. 2007 Jun 1;13(17):1741–50.
- Thijssen DHJ, Black MA, Pyke KE, Padilla J, Atkinson G, Harris RA, et al. Assessment of flow-mediated dilation in humans: a methodological and physiological guideline. American Journal of Physiology Heart and Circulatory Physiology [Internet]. 2011 Jan 1;300(1):H2-12. Available from: https://pubmed.ncbi.nlm.nih.gov/20952670/
- Ahn Y, Aung N, Ahn HS. A Comprehensive Review of Clinical Studies Applying Flow-Mediated Dilation. Diagnostics. 2024 Nov 8;14(22):2499–9.
- Masaki N, Higashimura Y, Takase B. Improvement of Cardiovascular Prognostic Value of Endothelial Function Tests by Repeated Measurements. Circulation Reports [Internet]. 2025 Jun 2 [cited 2026 Jan 5];7(7):568–77. Available from: https://www.jstage.jst.go.jp/article/circrep/7/7/7_CR-25-0042/_html/-char/en
- Caussy C, Reeder SB, Sirlin CB, Loomba R. Noninvasive, Quantitative Assessment of Liver Fat by MRI-PDFF as an Endpoint in NASH Trials. Hepatology. 2018 Aug;68(2):763–72.
- Mućka S, Miodońska M, Jakubiak GK, Starzak M, Cieślar G, Stanek A. Endothelial Function Assessment by Flow-Mediated Dilation Method: A Valuable Tool in the Evaluation of the Cardiovascular System. International Journal of Environmental Research and Public Health. 2022 Sep 7;19(18):11242.
- Tamaki N, Ajmera V, Loomba R. Non-invasive methods for imaging hepatic steatosis and their clinical importance in NAFLD. Nature Reviews Endocrinology. 2021 Nov 23;18(1):55–66.
- ZAKAI NA, KATZ R, JENNY NS, PSATY BM, REINER AP, SCHWARTZ SM, et al. Inflammation and hemostasis biomarkers and cardiovascular risk in the elderly: the Cardiovascular Health Study. Journal of Thrombosis and Haemostasis. 2007 Jun;5(6):1128–35.
- Tamaki N, Nagambika Munaganuru, Jung J, Yonan AQ, Loomba RR, Bettencourt R, et al. Clinical utility of 30% relative decline in MRI-PDFF in predicting fibrosis regression in non-alcoholic fatty liver disease. Gut. 2021 Apr 21;71(5):983–90.
- Upadhyay RK. Emerging Risk Biomarkers in Cardiovascular Diseases and Disorders. Journal of Lipids [Internet]. 2015;2015:1–50. Available from: https://www.hindawi.com/journals/jl/2015/971453/
- Al-Qaisi M. Measurement of endothelial function and its clinical utility for cardiovascular risk. Vascular Health and Risk Management. 2008 Jun;Volume 4:647–52.